
DNA double strand break repair and human disease
Research areas:
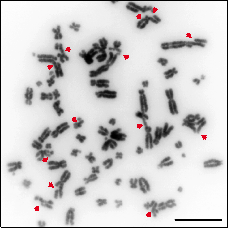 Chromosome breaks, commonly known as DNA Double Strand Breaks (DSBs) are the most cytotoxic genetic lesion known to man. Most usually, unrepaired DSB leads to cell death and for that reason their repair is essential for normal development. While the complete inability to repair chromosome breaks leads to embryonic lethality and cell death, mutations that hamper this repair lead to an increase on genomic instability, a driving force in cancer development and the cause of several rare diseases. Thus, defects in DSB repair cause genetically inherited syndromes, with or without cancer predisposition. The phenotypes associated with these syndromes are extremely varied, and can include growth and mental retardation, ataxia, skeletal abnormalities, immunodeficiency, premature aging, etc.
Chromosome breaks, commonly known as DNA Double Strand Breaks (DSBs) are the most cytotoxic genetic lesion known to man. Most usually, unrepaired DSB leads to cell death and for that reason their repair is essential for normal development. While the complete inability to repair chromosome breaks leads to embryonic lethality and cell death, mutations that hamper this repair lead to an increase on genomic instability, a driving force in cancer development and the cause of several rare diseases. Thus, defects in DSB repair cause genetically inherited syndromes, with or without cancer predisposition. The phenotypes associated with these syndromes are extremely varied, and can include growth and mental retardation, ataxia, skeletal abnormalities, immunodeficiency, premature aging, etc.
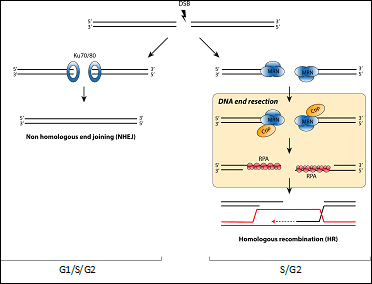
Chromosome breaks are repaired by two major mechanisms that compete for the same substrate. Both ends of the DSB can be simple re-joined with little or no processing, a mechanism known as non-homologous end-joining (NHEJ). On the other hand, DSBs can be processed and engaged in a more complex repair pathway called Homologous Recombination (HR). This pathway uses the information present in a homologue sequence. The balance between these two pathways is exquisitely controlled and its alteration leads to the appearance of chromosomal abnormalities and contribute to the diseases aforementioned. However, and despite its importance, the network controlling the choice between both is poorly understood. A critical step in the decision between both repair pathways is DNA end resection, a 5’-3’ degradation of one strand to create single stranded DNA. It is considered the key element in the decision between HR and NHEJ, as resected DNA is the substrate of recombination machinery and, more importantly, resected DNA effectively block NHEJ.
In our laboratory we are currently pursuing several research lines designed to investigate how the choice between both DSBs repair pathways is made, its relevance for cellular and organismal survival and disease, and its potential as a therapeutic target for the treatment of cancer and some genetically inherited disorders. This research lines can be divided in two main categories:
1. Detailed characterization of the role of CtIP in homologous recombination.
A key factor on the DSB repair choice is CtIP, a multifunctional protein that integrate multiple cellular signals. Moreover, we discovered that some CtIP mutations cause a Seckel-like syndrome, a genetically inherited dwarfism, Jawad syndrome and that CtIP is lost in aggressive breast cancer. In terms of DSB repair, CtIP act as a molecular switch that activates homologous recombination by activating the DNA resection step. Despite the importance of CtIP in this licensing step, we still not know how it acts molecularly. Thus, some of our efforts are set in characterize the molecular roles of CtIP and its regulation.
2. Global regulation of the balance between NHEJ and HR : relevance in cancer development and treatment.
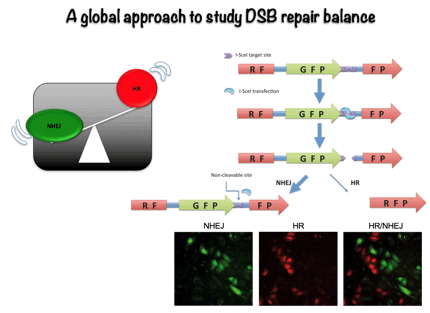
Most studies have traditionally focus in a specific mechanism of DNA repair, either NHEJ or HR. However, more recently it has become evident than miss regulation in the choice between different repair pathways might have stronger consequences for higher eukaryotes than simply blocking DSB repair. Thus, we have decided to study how the cell exerts the regulation between both repair types. To do so, we employed genomic approaches to try to find out the relevant components in this regulatory network using an easy, fluorescence based, assay to measure the ratio between NHEJ and HR. This way, we have discovered more than 300 new factors that control the choice between the different DSBs repair pathways. Currently, we are analyzing the role of those new factors in DSB repair pathway choice, mainly at the level of DNA resection licensing.
If you want to apply to our lab as a Master Student, PhD Students or Postdoc a send motivation letter, CV and contact details to pablo.huertas@cabimer.es. Postdoc applicant should include also the contact details of at least two referees.
Publications:
ORCID ID: https://orcid.org/0000-0002-1756-4449
RESEARCHER ID: http://www.researcherid.com/rid/N-2245-2016
SCOPUS ID: https://www.scopus.com/authid/detail.uri?authorId=6602760682
Lab Publications
1. EXO1 and DNA2-mediated ssDNA gap
expansion is essential for ATR activation and to maintain viability in BRCA1-deficient cells. García-Rodríguez N, Domínguez-García I,
Domínguez-Pérez MDC, Huertas P. Nucleic Acids Res. 2024 Jun
24;52(11):6376-6391. doi: 10.1093/nar/gkae317.
2. Centriolar subdistal appendages promote double-strand break repair through homologous recombination. Rodríguez-Real G, Domínguez-Calvo A, Prados-Carvajal R, Bayona-Feliú A, Gomes-Pereira S, Balestra FR, Huertas P. (Author 7/7) EMBO Rep. 2023 Oct 9;24(10):e56724. doi: 10.15252/embr.202256724.
3. ADAR2-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Jimeno S, Prados-Carvajal R, Fernández-Ávila MJ, et al, Huertas P. (Author 17/17). Nature Communication, Volume 12, Issue 1December 2021 Article number 5512
4. CtIP -mediated alternative mRNA splicing finetunes the DNA damage response. Prados-Carvajal R, Rodriguez-Real G, Gutierrez-Pozo G, Huertas P. (Author 4/4) RNA. 2020 Dec 9:rna.078519.120. doi: 10.1261/rna.078519.120
5. Methylation of the central transcriptional regulator KLF4 by PRMT5 is required for DNA end resection and recombination. Checa-Rodríguez C, Cepeda-García C, Ramón J, López-Saavedra A, Balestra FR, Domínguez-Sánchez MS, Gómez-Cabello D and Huertas P. (Author 8/8) DNA Repair (Amst). 2020 Oct;94:102902. doi: 10.1016/j.dnarep.2020.102902.
6. ALC1/eIF4A1-mediated regulation of CtIP mRNA stability controls DNA end resection. Mejías-Navarro F, Rodríguez-Real G, Ramón J, Camarillo R, Huertas P. (Author 5/5) PLoS Genet. 2020 May 11;16(5):e1008787. doi: 10.1371/journal.pgen.1008787.
7. The Helicase PIF1 Facilitates Resection over Sequences Prone to Forming G4 Structures. Jimeno, S., Camarillo, R., Mejías-Navarro, F., Fernández-Ávila, M.J., Soria-Bretones, I., Prados-Carvajal, R., Huertas, P. (Author 7/7) Cell Rep. 2018 Sep 18;24(12):3262-3273.e4. doi: 10.1016/j.celrep.2018.08.047.
Collaborations
8. The CDK12-BRCA1 signaling axis mediates
dinaciclib-associated radiosensitivity through p53-mediated cellular
senescence. Flores NG, Fernández-Aroca DM,
Garnés-García C, Domínguez-Calvo A, Jiménez-Suárez J, Sabater S,
Fernández-Aroca P, Andrés I, Cimas FJ, de Cárcer G, Belandia B, Palmero I,
Huertas P, Ruiz-Hidalgo MJ, Sánchez-Prieto R. Mol Oncol. 2024 Dec 3. doi:
10.1002/1878-0261.13773. Online ahead of print. PMID: 39626031
9. Mechanism of BRCA1-BARD1 function in DNA
end resection and DNA protection. Ceppi I, Dello Stritto MR, Mütze M,
Braunshier S, Mengoli V, Reginato G, Võ HMP, Jimeno S, Acharya A, Roy M,
Sanchez A, Halder S, Howard SM, Guérois R, Huertas P, Noordermeer SM, Seidel R,
Cejka P. Nature. 2024 Oct;634(8033):492-500. doi: 10.1038/s41586-024-07909-9.
Epub 2024 Sep 11. PMID: 39261728.
10. BRCA1/BARD1 ubiquitinates PCNA in
unperturbed conditions to promote continuous DNA synthesis. Salas-Lloret D, García-Rodríguez N,
Soto-Hidalgo E, González-Vinceiro L, Espejo-Serrano C, Giebel L, Mateos-Martín
ML, de Ru AH, van Veelen PA, Huertas P, Vertegaal ACO, González-Prieto R. Nat
Commun. 2024 May 20;15(1):4292. doi: 10.1038/s41467-024-48427-6. PMID: 38769345 .
11. N7-methylguanosine methylation of tRNAs
regulates survival to stress in cancer. García-Vílchez R, Añazco-Guenkova AM,
López J, Dietmann S, Tomé M, Jimeno S, Azkargorta M, Elortza F, Bárcena L,
Gonzalez-Lopez M, Aransay AM, Sánchez-Martín MA, Huertas P, Durán RV, Blanco S.
Oncogene. 2023 Oct;42(43):3169-3181. doi: 10.1038/s41388-023-02825-0. Epub 2023
Sep 2.
12. PLK1 regulates CtIP and DNA2 interplay in long-range DNA end resection. Ceppi I, Cannavo E, Bret H, Camarillo R, Vivalda F, Thakur RS, Romero-Franco A, Sartori AA, Huertas P, Guérois R, Cejka P. (Author 9/11) Genes Dev. 2023 Feb 1;37(3-4):119-135. doi: 10.1101/gad.349981.122.
13. EXOSC10 is required for RPA assembly and controlled DNA end resection at DNA double-strand breaks. Domingo-Prim J, et al (Author 7/8). Nat Commun. 2019 May 13;10(1):2135. doi: 10.1038/s41467-019-10153-9.
14. UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Jachimowicz, RD, et al (Author 24/31). Cell. 2018 Dec 21. doi: 10.1016/j.cell.2018.11.024.
15. Decapping protein EDC4 regulates DNA repair and phenocopies BRCA1. Hernández G, et al (Author 22/26). Nature Communications, 2018 9(1), 967.
Collaboration papers
1.
PLK1 regulates CtIP and DNA2 interplay in
long-range DNA end resection. Ceppi I, Cannavo E, Bret H, Camarillo R, Vivalda
F, Thakur RS, Romero-Franco A, Sartori AA, Huertas P, Guérois R, Cejka
P. (Author 9/11) Genes Dev. 2023 Feb 1;37(3-4):119-135. doi:
10.1101/gad.349981.122.
2.
EXOSC10 is required for RPA assembly and controlled
DNA end resection at DNA double-strand breaks. Domingo-Prim J, et al (Author
7/8). Nat Commun. 2019 May 13;10(1):2135. doi: 10.1038/s41467-019-10153-9.
3.
UBQLN4 represses homologous recombination and is
overexpressed in aggressive tumors. Jachimowicz, RD, et al (Author 24/31).
Cell. 2018 Dec 21. doi: 10.1016/j.cell.2018.11.024.
4.
Decapping
protein EDC4 regulates DNA repair and phenocopies BRCA1. Hernández G, et al (Author
22/26). Nature Communications,
2018 9(1), 967.

 Dr. Pablo Huertas Sánchez
Dr. Pablo Huertas Sánchez
- Maikel C. Pozo
- Dr. Younes El Yousfi El Mourabit
- Dr. María Jesús Fernández Ávila
- Dr. Laura Dolores Gallego Valle
- Dr. Pedro Ortega Moreno
- Dr. María Rosario Prados Carvajal
- Candela Caballero Fernández
- Nieves Iria Domínguez García
- Lucía González López
- María Isabel Jiménez López
- Amador Romero Franco
- Mónica del Pilar Perales Viedma
- Natalia Quesada Lechón
- Marina De Castro Tarragó
- Irene García Pérez



