
Señalización en la muerte celular
Regulación de la apoptosis en células normales y tumorales
En los últimos años ha tenido lugar un enorme progreso en la comprensión de la señalización implicada en la muerte de células tumorales por la activación de los receptores del ligando de muerte celular TRAIL. Además, la observación inicial de que TRAIL induce específicamente la muerte celular en las células tumorales, sin afectar a células normales, ha inspirado numerosos ensayos clínicos con agentes terapéuticos diseñados para activar los receptores proapoptóticos de TRAIL. A pesar de estos avances, todavía quedan muchas incógnitas sin aclarar acerca de los mecanismos que subyacen a la resistencia de las células normales a TRAIL. Por otra parte, a pesar de los prometedores resultados iniciales, un número de células tumorales, como los de mama, páncreas, melanoma y neuroblastoma son, con frecuencia, resistentes a la apoptosis inducida por TRAIL. Sin embargo, un consenso general para explicar esta resistencia a TRAIL aún no ha sido alcanzado.
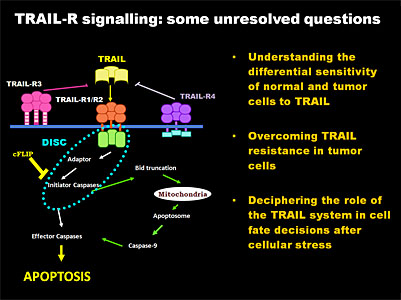
Nuestro grupo ha venido investigando diferentes aspectos de la regulación de la apoptosis desde 1989, con proyectos de investigación de diversas fuentes y publicaciones en este campo. Recientemente nuestro grupo ha demostrado que TRAIL induce una autofagia citoprotectora en células humanas epiteliales de mama no tumorales. Esta autofagia está mediada por la activación de la proteína quinasa TAK1 y es, a su vez, dependiente de la proteína quinasa activada por AMP (AMPK) que inhibe al complejo con actividad quinasa mTORC1, un potente inhibidor de la autofagia. Estos hallazgos identifican TAK1 como un activador de la AMPK y la autofagia citoprotectora y de ese modo como un regulador de la homeostasis de la energía celular y la supervivencia. En este tema, el objetivo general de nuestra investigación actual es dilucidar los mecanismos moleculares que regulan la apoptosis inducida por TRAIL y la autofagia en las células epiteliales de mama y de caracterizar las vías de señalización que generan resistencia a TRAIL en las células tumorales .
Un proceso fundamental en la progresión de carcinomas es la adquisición de un fenotipo mesenquimal y la pérdida de características epiteliales. Esta transición epitelio-mesénquima (EMT) facilita la adquisición de propiedades invasivas y aumenta la resistencia a procesos de apoptosis y senescencia. Sin embargo, los mecanismos moleculares responsables de la regulación de la sensibilidad a TRAIL durante la EMT son actualmente desconocidos. En este punto, es importante mencionar la relación entre la señalización de EMT y las alteraciones en la polaridad ápico-basal que tienen lugar en las etapas tempranas del proceso tumoral, de forma que inhibidores transcripcionales de la expresión de E-cadherina durante la EMT, suprimen tambien la expresión de proteínas relevantes de los complejos de polaridad. Finalmente, es importante resaltar que en modelos tridimensionales de células epiteliales de mama, la pérdida de polaridad ápico-basal resulta en una mayor sensibilidad a diversos estímulos apoptóticos, entre los que se incluye a TRAIL. Por lo tanto, dado que TRAIL y anticuerpos agonistas de receptores de TRAIL están actualmente en ensayos clínicos como antitumorales, es de gran importancia conocer las bases moleculares de la sensibilidad a TRAIL durante la EMT para optimizar estas estrategias terapéuticas. En este sentido, tenemos la intención de investigar el papel de la FLIP en la regulación de la resistencia de TRAIL en EMT y, en particular, durante la progresión del tumor.
La inhibición farmacológica de la salida de mitosis es una estrategia de terapia antitumoral que recientemente ha sido propuesta por diversos grupos, basada en el modelo de dos “competing networks”, la degradación de ciclina B y la acumulación de señales proapoptóticas, para explicar el destino de células tumorales paradas en mitosis. En este sentido, nuestros resultados recientes indican que la parada mitótica induce la degradación por el proteasoma del inhibidor de la ruta extrínseca FLIP y sensibiliza a las células tumorales a TRAIL. Nuestro objetivo futuro es entender la regulación de la expresión FLIP durante la mitosis y el papel de la quinasa dependiente de ciclina B1 (CDK1) en el control de los niveles de FLIP y la sensibilidad de TRAIL.
El microambiente tumoral se caracteriza por la escasez de nutrientes, la acidosis y una hipoxia severa. Estos factores combinados provocan la acumulación de proteínas mal plegadas en el retículo endoplasmático (RE), lo que induce la activación de una ruta de señalización denominada UPR (unfolded protein response) para facilitar la supervivencia y el crecimiento tumoral. Sin embargo, en determinadas condiciones, la activación de la UPR en tumores por señales microambientales puede modificar la respuesta adaptativa y favorecer la expresión de las proteínas de la vía extrínseca de la apoptosis, aumentando la sensibilidad de estas células a TRAIL. Nuestros resultados demuestran que en células epiteliales de mama que expresan formas constitutivamente activas del oncogen ErbB2, los tratamientos que activan la UPR inducen la expresión del receptor proapoptótico de TRAIL, DR5/TRAIL-R2, disminuyen la expresión de proteínas antiapoptóticas como FLIP y activan la apoptosis a través de la ruta extrínseca. Actualmente estamos tratando de identificar las ramas de la UPR que están involucrados en la regulación de estas proteínas y los mecanismos que controlan el paso de una respuesta adaptativa al estrés en el RE a una respuesta apoptótica, en células tumorales.
Financiación
- Ministerio de Economía y Competitividad (SAF2015-64383-P)
- Proyecto de Excelencia de la Junta de Andalucía (P12-BIO-0778)
- Centro de Investigación Biomédica en Red de Cáncer (CIBERONC) (CB16/12/00421)
- Red de Excelencia para el estudio de la Autofagia (BFU2015-71869-REDT)
- Unión Europea (FEDER)
Publicaciones seleccionadas:
C. Ruiz-Ruiz, G. Robledo, J. Font, M. Izquierdo and A. López- Rivas. Protein Kinase C inhibits CD95(Fas/APO-1)-mediated apoptosis by at least two different mechanisms in Jurkat T cells. J. Immunol. (1999) 163: 4737-4746
C. Ruiz-Ruiz and A. López-Rivas. p53-mediated up-regulation of CD95 is not involved in genotoxic drug-induced apoptosis of human breast tumor cells. Cell Death and Differentiation (1999) 6, 271-280
Ruiz-Ruiz, C. Muñoz-Pinedo and A. López-Rivas. Interferon-gamma treatment elevates caspase-8 expression and sensitises human breast tumor cells to a death receptor-induced mitochondria-operated apoptotic program. Cancer Res. (2000) 60, 5673-5680
Sarker, C. Ruiz-Ruiz and A. López-Rivas. Activation of protein kinase C inhibits TRAIL-induced caspases activation, mitochondrial events and apoptosis in a human leukemic T cell line. Cell Death and Differentiation (2001) 8: 172-181
Muñoz-Pinedo, C. Ruiz-Ruiz, C. Ruiz de Almodovar, C. Palacios and A. López-Rivas. Inhibition of glucose metabolism sensitises tumor cells to death receptor-triggered apoptosis through enhancement of DISC formation and apical procaspase-8 processing. J. Biol. Chem. (2003) 278: 12759-12768
Ruiz de Almodovar, C. Ruiz-Ruiz, A. Rodríguez, G. Ortiz-Ferrón, J.M. Redondo and A. López-Rivas. TRAIL decoy receptor TRAIL-R3 is upregulated by p53 in breast tumor cells through a mechanism involving an intronic p53 binding site. J. Biol. Chem. (2004) 279: 4093-4101
Ruiz-Ruiz, C. Ruiz de Almodóvar, A. Rodríguez, G. Ortiz-Ferrón, J.M. Redondo and A. López-Rivas. The up-regulation of human caspase-8 by interferon-gamma in breast tumor cells requires the induction and action of the transcription factor IRF-1. J. Biol. Chem. (2004) 279:19712-19720
Palacios, R. Yerbes and A. López-Rivas. Flavopiridol induces cFLIP degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Research (2006) 66, 8858-8869
Gustavo Ortiz-Ferrón, Stephen W. Tait, Gema Robledo, Evert de Vries, Jannie Borst, and Abelardo López-Rivas. The mitogen-activated protein kinase pathway can inhibit TRAIL-induced apoptosis by prohibiting association of truncated Bid with mitochondria. Cell Death and Differentiation (2006) 13, 1857-1865
Griselda Herrero-Martín, Maria Høyer-Hansen, Celina García-García, Claudia Fumarola, Lone Batholm, Thomas Farkas, Abelardo López-Rivas@ and Marja Jäättelä@. TAK1 activates AMPK and AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. The EMBO Journal (2009) 28, 677-685
Tania Sánchez-Pérez, Gustavo Ortiz-Ferrón and Abelardo López-Rivas “Mitotic arrest and JNK-induced proteasomal degradation of FLIP and Mcl-1 are key event in the sensitization of breast tumor cells to TRAIL by anti-microtubule agents” Cell Death and Differentiation (2010) 17:883-94.
Carmen Palacios, Ana Isabel López-Pérez and Abelardo López-Rivas. Down-regulation of RIP expression by 17-Dimethylaminoethylamino-17-Demethoxygeldanamycin promotes TRAIL-induced apoptosis in breast tumor cells. Cancer Letters (2010) 287, 207-215.
Rosario Yerbes, Carmen Palacios, Mauricio J. Reginato and Abelardo López-Rivas “Cellular FLIPL plays a survival role and regulates morphogenesis in breast epithelial cells” BBA Mol Cell Res (2011) 1813, 168-178.
José Manuel Rodríguez-Vargas, María José Ruiz-Magaña, Carmen Ruiz-Ruiz, José Antonio Muñoz-Gámez, Eva Siles, Abelardo López Rivas, Marja Jaattela, and F. Javier Oliver. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Research (2012) 22: 1181-1198
Rosario Yerbes, Abelardo López-Rivas*, Mauricio J. Reginato and Carmen Palacios*. Control of FLIPL expression and TRAIL resistance by the extracellular signal-regulated kinase (ERK)1/2 pathway in breast epithelial cells. Cell Death and Differentiation (2012) 19: 1908-1916.
Rosa Martín-Pérez, Carmen Palacios*, Rosario Yerbes*, Ana Cano-González, Daniel Iglesias-Serret, Joan Gil, Mauricio J. Reginato and Abelardo López-Rivas. (*equal contribution) Activated HER2 licenses sensitivity to apoptosis upon endoplasmic reticulum stress through a PERK-dependent pathway. Cancer Research (2014) 74: 1766-1777
Tania Sánchez-Pérez, René H. Medema and Abelardo López-Rivas. Delaying mitotic exit down-regulates FLIP expression and strongly sensitizes tumor cells to TRAIL. Oncogene (2015) 34: 661-669
Katiuska González-Arzola, Irene Díaz-Moreno, Ana Cano-González, Antonio Díaz-Quintana, Adrián Velázquez-Campoy, Blas Moreno-Beltrán, Abelardo López-Rivas and Miguel Á. de la Rosa. Structural Basis for Inhibition of the Histone Chaperone Activity of SET/TAF-Ib by Cytochrome c. PNAS (2015) 112: 9908-9913
Ana Cano-González and Abelardo López-Rivas. Opposing roles of TGF-b and EGF in the regulation of TRAIL-induced apoptosis in human breast epithelial cells. BBA Mol Cell Res (2016) 1863: 2104-2114
Rosa Martín-Pérez, Rosario Yerbes, Rocío Mora-Molina, Ana Cano-González, Joaquín Arribas, Massimiliano Mazzone, Abelardo López-Rivas@ and Carmen Palacios@. Oncogenic p95Her2/611CTF primes human breast epithelial cells for metabolic stress-induced activation of TRAIL-R/Caspase-8-dependent apoptosis.Oncotarget (2017) 8:93688-93703
Cristina Muñoz-Pinedo and Abelardo López-Rivas. A role for caspase-8 and TRAIL-R2/DR5 in ER-stress induced apoptosis. Cell Death and Differentiation (2018) (doi: 10.1038/cdd.2017.155.)
Ana Cano-González, Marta Mauro-Lizcano, Daniel Iglesias-Serret, Joan Gil and Abelardo López-Rivas. Involvement of both caspase-8 and Noxa-activated pathways in ER stress-induced apoptosis in triple-negative breast tumor cells. Cell Death and Disease (2018) 9(2):134. doi: 10.1038/s41419-017-0164-7.
Marta Mauro-Lizcano and Abelardo López-Rivas. Glutamine metabolism regulates FLIP expression and sensitivity to TRAIL in triple-negative breast cancer cells. Cell Death and Disease (2018) 9(2):205. doi: 10.1038/s41419-018-0263-0.

-
 Dr. Abelardo López Rivas
Dr. Abelardo López Rivas
- Dr. Carmen Palacios Casanova
- Dr. Rosario Yerbes Cadenas
- Younes El Yousfi El Mourabit
- Rocío Mora Molina
- Francisco Javier Fernández Farrán



