RNA Metabolism and DNA Repair in Cancer Therapy Resistance
e-mail: rosario.prados@cabimer.es
ORCID: 0000-0003-4721-6311
X: @marpracar
Linkedin: https://www.linkedin.com/in/rosario-prados-phd-53a587106/

RNA Metabolism and DNA Repair in Cancer Therapy Resistance
e-mail: rosario.prados@cabimer.es
ORCID: 0000-0003-4721-6311
X: @marpracar
Linkedin: https://www.linkedin.com/in/rosario-prados-phd-53a587106/

Overview
Cancer cells rewire RNA metabolism to survive genotoxic stress and escape treatment, and this rewiring can also be turned against them. Our research focuses on the crosstalk between RNA metabolism and the DNA damage response, and on how manipulating this crosstalk can revert drug resistance in cancer. We study the molecular determinants that shift a cancer cell from resistance back to sensitivity toward DNA-damaging agents and targeted inhibitors, with the long-term goal of defining combination strategies that can resensitize resistant tumors in the clinic.
Current research lines
1. Determinants of PARP inhibitor (PARPi) resistance in BRCA-deficient cancers
PARP inhibitors exploit a concept known as synthetic lethality: tumor cells deficient in homologous recombination (HR) rely on PARP enzymes to cope with the DNA damage that accumulates when this repair pathway is not available. Blocking PARP activity overwhelms these cells with unrepaired damage that they cannot resolve, while cells with intact HR remain largely unaffected (Figure 1). This selectivity has transformed the treatment of breast, ovarian, pancreatic and prostate cancers with HR deficiencies. However, tumors frequently find ways to restore their ability to tolerate DNA damage, and acquired resistance to PARP inhibitors remains one of the main obstacles to durable responses in the clinic.
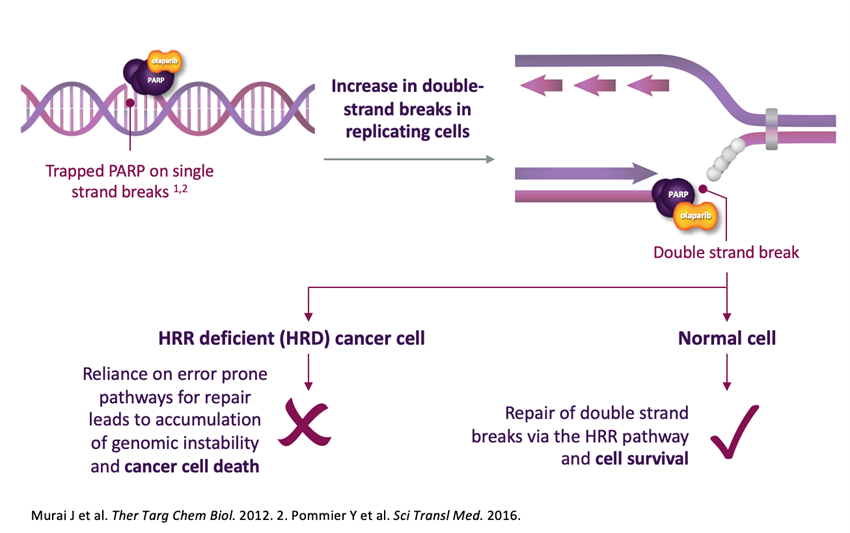
Figure 1. Mechanism of PARP inhibitor action in HR-deficient cancer cells. PARP inhibitors such as olaparib trap PARP on single-strand DNA breaks, preventing its release and increasing the load of double-strand breaks encountered during replication. In HR-deficient (HRD) cells, these breaks cannot be repaired accurately and instead accumulate through error-prone pathways, driving genomic instability and cancer cell death. In cells with intact HR, the same breaks are efficiently resolved and cell survival is preserved.
Our work explores how factors involved in RNA metabolism shape the balance between sensitivity and resistance to PARP inhibitors in BRCA1/2-deficient cancer cells, where BRCA proteins are central to HR. We are investigating how these factors affect the way cells handle DNA damage and the signaling pathways that connect DNA repair to cell survival, with the aim of identifying vulnerabilities that can be exploited pharmacologically. One of the central determinants we are studying is the ADAR family of RNA-editing enzymes, whose activity influences how BRCA-deficient cells respond to DNA damage and to PARP inhibitor treatment. Our goal is to define how modulating RNA-metabolism pathways, can help resensitize resistant, BRCA-deficient tumors to PARP inhibitors, and to translate this into rational combination strategies.
2. Reverting resistance to KRAS-targeted therapy in lung and pancreatic cancer models
Oncogenic KRAS is one of the most frequent drivers of lung and pancreatic cancer, and resistance to KRAS-targeted therapies is a growing clinical problem. RNA-metabolism factors determine how oncogene-driven tumors respond to KRASG12C inhibitors. In resistant non-small cell lung cancer (NSCLC) and pancreatic ductal adenocarcinoma (PDAC) models, depletion of specific RNA-editing enzymes bidirectionally alters KRAS mRNA and protein levels through post-transcriptional mechanisms. This project aims to exploit these mechanisms as entry points to resensitize resistant tumors to KRAS-targeted therapy.
Projects as Principal Investigator:
Projects as Researcher:
A complete list can be obtained at https://orcid.org/my-orcid?orcid=0000-0003-4721-6311



