
Retinal neurodegeneration and advanced therapies
Research lines:
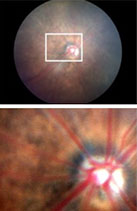 Degenerative diseases of the retina are a heterogeneous group of ophthalmological pathologies, which are produced by the degeneration of photoreceptors and/or retinal pigment epithelium, with the consequent progressive loss of visual acuity leading to blindness. According to statistics published by the World Health Organization (WHO), it is estimated that at least 2.2 billion people have visual impairment or blindness, of which at least 1 billion have a visual impairment that could have been prevented or even treated. With the recent development of advanced therapies in ophthalmic clinical practice, some cases of retinal degeneration, which were previously incurable, are currently being treated. For this reason, more research is needed to continue the worldwide fight against blindness.
Degenerative diseases of the retina are a heterogeneous group of ophthalmological pathologies, which are produced by the degeneration of photoreceptors and/or retinal pigment epithelium, with the consequent progressive loss of visual acuity leading to blindness. According to statistics published by the World Health Organization (WHO), it is estimated that at least 2.2 billion people have visual impairment or blindness, of which at least 1 billion have a visual impairment that could have been prevented or even treated. With the recent development of advanced therapies in ophthalmic clinical practice, some cases of retinal degeneration, which were previously incurable, are currently being treated. For this reason, more research is needed to continue the worldwide fight against blindness.
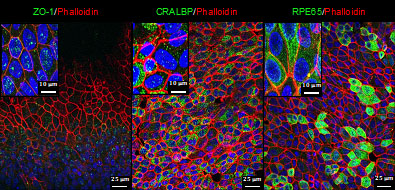
Our research focuses on inherited monogenic retinal dystrophies such as retinitis pigmentosa (RP) and genetically complex diseases affecting vision like age-related macular degeneration (AMD). Starting from the genetic information, we pursue the study of specific disease mechanisms to explore and test new therapeutics for degenerative retinopathies that are currently incurable. To study the molecular mechanisms of retinal degeneration, we used both classical in vivo models of retinal degeneration and cellular models by reprogramming induced pluripotent stem (iPS) cells obtained from clinical samples and their subsequent differentiation into specific cell types of the retina. All our experimental tools and human resources are ultimately focused on translational research since the final goal of our research project is the development and preclinical evaluation of new therapies to treat retinal degenerative diseases. Our approach includes gene therapy and gene editing using CRISPR/Cas9 technology, cell therapy using iPS, development of new neuroprotective small molecules as well as the application of nanotechnology and biomaterials for therapeutic purposes. In the frame of translational research, we have created the Spin-off Limno Pharma S.L. to boost a future clinical trial for RP and AMD. We also work in close collaboration with the Clinical Ophthalmology Department of University Hospital Virgen Macarena and patient’s associations.
The main research lines of the group are:
i) Functional characterization of genes related to eye disease.
ii) Impact of mutations on protein function to understand the molecular basis of the disease.
iii)Development of novel therapeutic approaches and biomarkers for retinal degenerative diseases.
Current research projects:
- Preclinical study to evaluate the safety and efficacy of retinal pigment epithelium transplants grown on a nanostructured hydrogel to treat age-related macular degeneration (AMD CELLS-II). PI20/00043
- Preclinical study of ex vivo genomic editing therapy for retinitis pigmentosa. PI-0099-2018
- Preclinical study to evaluate SIRT1 and VEGF modulating drugs as a therapeutic for macular degeneration and diabetic retinopathy (SIRT-IN-EYES). PI17/01026
Predoctoral and Master’s students are welcome to join our research lines.
Recent Funding:

Scientific production (last 5 years)
- Caballano Infantes E, Clauzon L, de la Cerda Haynes B, Díaz-Corrales F. Generation of the human iPSC line ESi132-A from a patient with retinitis pigmentosa caused by a mutation in the PRPF31 gene. Stem Cell Res. 2025 Feb;82:103623. doi: 10.1016/j.scr.2024.103623. PMID: 39755010.
- Valdés-Sánchez L, Moshtaghion SM, Caballano-Infantes E, Peñalver P, Rodríguez-Ruiz R, González-Alfonso JL, Plou FJ, Desmet T, Morales JC, Díaz-Corrales FJ. Synthesis and evaluation of glucosyl-, acyl- and silyl-resveratrol derivatives as retinoprotective agents: Piceid octanoate notably delays photoreceptor degeneration in a retinitis pigmentosa mouse model. Pharmaceuticals (Basel). 2024 Nov 5;17(11):1482. doi: 10.3390/ph17111482. PMID: 39598393.
- Moshtaghion SM, Caballano-Infantes E, Plaza Reyes Á, Valdés-Sánchez L, Fernández PG, de la Cerda B, Riga MS, Álvarez-Dolado M, Peñalver P, Morales JC, Díaz-Corrales FJ. Piceid octanoate protects retinal cells against oxidative damage by regulating the sirtuin 1/poly-ADP-ribose polymerase 1 axis in vitro and in rd10 mice. Antioxidants (Basel). 2024 Feb 4;13(2):201. doi: 10.3390/antiox13020201. PMID: 38397799.
- Valdés-Sánchez L, Borrego-González S, Montero-Sánchez A, Massalini S, de la Cerda B, Díaz-Cuenca A, Díaz-Corrales FJ. Mesoporous silica-based nanoparticles as non-viral gene delivery platform for treating retinitis pigmentosa. J Clin Med. 2022 Apr 13;11(8):2170. doi: 10.3390/jcm11082170. PMID: 35456263.
- Urbano-Gámez JD, Valdés-Sánchez L, Aracil C, de la Cerda B, Perdigones F, Plaza Reyes Á, Díaz-Corrales FJ, Relimpio López I, Quero JM. Biocompatibility study of a commercial printed circuit board for biomedical applications: Lab-on-PCB for organotypic retina cultures. Micromachines (Basel). 2021 Nov 29;12(12):1469. doi: 10.3390/mi12121469. PMID: 34945319.
- Buono L, Corbacho J, Naranjo S, Almuedo-Castillo M, Moreno-Marmol T, de la Cerda B, Sanabria-Reinoso E, Polvillo R, Díaz-Corrales FJ, Bogdanovic O, Bovolenta P, Martínez-Morales JR. Analysis of gene network bifurcation during optic cup morphogenesis in zebrafish. Nat Commun. 2021 Jun 23;12(1):3866. doi: 10.1038/s41467-021-24169-7. Erratum in: Nat Commun. 2021 Jul 27;12(1):4665. doi: 10.1038/s41467-021-24834-x. PMID: 34162866.
- Cañibano-Hernández A, Valdes-Sanchez L, Garcia-Delgado AB, Ponte-Zúñiga B, Diaz-Corrales FJ, de la Cerda B. Generation of the human iPSC line ESi082-A from a patient with macular dystrophy associated to mutations in the CRB1 gene. Stem Cell Res. 2021 May;53:102301. doi: 10.1016/j.scr.2021.102301. PMID: 33773389.
- Borrego-González S, de la Cerda B, Díaz-Corrales FJ, Díaz-Cuenca A. Nanofibrous matrix of defined composition sustains human induced pluripotent stem cell culture. ACS Appl Bio Mater. 2021 Apr 19;4(4):3035-3040. doi: 10.1021/acsabm.0c00425. PMID: 35014391.
- Ajana S, Cougnard-Grégoire A, Colijn JM, Merle BMJ, Verzijden T, de Jong PTVM, et al.; EYE-RISK Consortium. Predicting progression to advanced age-related macular degeneration from clinical, genetic, and lifestyle factors using machine learning. Ophthalmology. 2021 Apr;128(4):587-597. doi: 10.1016/j.ophtha.2020.08.031. PMID: 32890546.
- Acar İE, Lores-Motta L, Colijn JM, Meester-Smoor MA, Verzijden T, Cougnard-Gregoire A, et al.; EYE-RISK Consortium. Integrating metabolomics, genomics, and disease pathways in age-related macular degeneration: The EYE-RISK Consortium. Ophthalmology. 2020 Dec;127(12):1693-1709. doi: 10.1016/j.ophtha.2020.06.020. Epub 2020 Jun 14. PMID: 32553749.
- Valdés-Sánchez L, Calado SM, De la Cerda B, Aramburu A, García-Delgado AB, Massalini S, Montero-Sánchez A, Bhatia V, Rodríguez-Bocanegra E, Diez-Lloret A, Rodríguez-Martínez D, Chakarova C, Bhattacharya SS, Díaz-Corrales FJ. Retinal pigment epithelium degeneration caused by aggregation of PRPF31 and the role of HSP70 family of proteins. Mol Med, 2019. Dec 31;26(1). doi: 10.1186/s10020-019-0124-z
- Garcia-Delgado AB, Calado SM, Valdes-Sanchez LM, Montero-Sanchez A, Ponte-Zuñiga B, De la Cerda B, Bhattacharya SS, Diaz-Corrales FJ. Generation of a human iPS cell line (CABi003-A) from a patient with age-related macular degeneration carrying the CFH Y402H polymorphism. Stem Cell Res, 2019. doi: 10.1016/j.scr.2019.101473. 38:101473.
- Kersten E, Dammeier S, Ajana S, Groenewoud JMM, Codrea M, Klose F, Lechanteur YT, Fauser S, Ueffing M, Delcourt C, Hoyng CB, de Jong EK, den Hollander AI; EYE-RISK Consortium. Metabolomics in serum of patients with non-advanced age-related macular degeneration reveals aberrations in the glutamine pathway. PLoS One, 0218457, 14(6). 2019. doi: 10.1371/journal.pone.
- Cabello M, Mozo M, De la Cerda B, Aracil C, Díaz-Corrales FJ, Perdigones F, Valdés-Sánchez L, Relimpio I, Bhattacharya SS, Quero JM. Electrostimulation in an autonomous culture lab-on-chip provides neuroprotection of a retinal explant from a retinitis pigmentosa mouse-model. Sens Actuators B Chem. 2019;288:337–46. doi:10.1016/j.snb.2019.02.118.
- García Delgado AB, de la Cerda B, Alba Amador J, Valdés Sánchez ML, Fernández-Muñoz B, Relimpio López I, Rodríguez de la Rúa E, Díez Lloret A, Calado SM, Sánchez Pernaute R, Bhattacharya SS, Díaz Corrales FJ. Subretinal transplant of induced pluripotent stem cell-derived retinal pigment epithelium on nanostructured fibrin-agarose. Tissue Eng Part A. 2019 May;25(9-10):799-808. doi: 10.1089/ten.TEA.2019.0007. PMID: 30963803.
- de la Cerda B, Díez-Lloret A, Ponte B, Vallés-Saiz L, Calado SM, Rodríguez-Bocanegra E, Garcia-Delgado AB, Moya-Molina M, Bhattacharya SS, Díaz-Corrales FJ. Generation and characterization of the human iPSC line CABi001-A from a patient with retinitis pigmentosa caused by a novel mutation in PRPF31 gene. Stem Cell Res. 2019 Apr;36:101426. doi: 10.1016/j.scr.2019.101426. PMID: 30921587.
- Colijn JM, den Hollander AI, Demirkan A, Cougnard-Grégoire A, Verzijden T, Kersten E, et al.; EYE-RISK Consortium. Increased high-density lipoprotein levels associated with age-related macular degeneration: Evidence from the EYE-RISK and European Eye Epidemiology Consortia. Ophthalmology. 2019 Mar;126(3):393-406. doi: 10.1016/j.ophtha.2018.09.045. PMID: 30315903.
- Merle BMJ, Colijn JM, Cougnard-Grégoire A, de Koning-Backus APM, Delyfer MN, Kiefte-de Jong JC, et al.; EYE-RISK Consortium. Mediterranean diet and incidence of advanced age-related macular degeneration: The EYE-RISK Consortium. Ophthalmology. 2019 Mar;126(3):381-390. doi: 10.1016/j.ophtha.2018.08.006. PMID: 30114418.
- Valdés-Sánchez L, García-Delgado AB, Montero-Sánchez A, de la Cerda B, Lucas R, Peñalver P, Morales JC, Bhattacharya SS, Díaz-Corrales FJ. The resveratrol prodrug JC19 delays retinal degeneration in rd10 mice. Adv Exp Med Biol. 2019;1185:457-462. doi: 10.1007/978-3-030-27378-1_75. PMID: 31884654.
- Calado SM, Garcia-Delgado AB, De la Cerda B, Ponte-Zuñiga B, Bhattacharya SS, Díaz-Corrales FJ. Generation of a human iPS cell line from a patient with retinitis pigmentosa due to EYS mutation. Stem Cell Res. 2018;33:251-4. doi: 10.1016/j.scr.2018.11.002. PMID: 30471616.
- Garcia-Delgado AB, Valdés-Sánchez L, Calado SM, Diaz-Corrales FJ*, Bhattacharya SS. Rasagiline delays retinal degeneration in a mouse model of retinitis pigmentosa via modulation of Bax/Bcl-2 expression. CNS Neurosci Ther. 2018;24(5):448-55. doi: 10.1111/cns.12805. PMID: 29372592.
- Colijn JM, Buitendijk GHS, Prokofyeva E, Alves D, Cachulo ML, Khawaja AP, et al.; EYE-RISK consortium; European Eye Epidemiology (E3) consortium. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology. 2017;124(12):1753-63. PMDI: 28712657.
- Pensado A, Diaz-Corrales FJ*, De la Cerda B, Valdés-Sánchez L, Del Boz AA, Rodriguez-Martinez D, et al. Span poly-L-arginine nanoparticles are efficient non-viral vectors for PRPF31 gene delivery: An approach of gene therapy to treat retinitis pigmentosa. Nanomedicine. 2016;12(8):2251-60. doi: 10.1016/j.nano.2016.06.007. PMID: 27381066.
- Calado SM, Diaz-Corrales F, Silva GA. pEPito-driven PEDF Expression Ameliorates Diabetic Retinopathy Hallmarks. Hum Gene Ther Methods. 2016;27(2):79-86. doi: 10.1089/hgtb.2015.169. PMID: 26942449.
Patents
- Berta De la Cerda Haynes; Francisco Javier Díaz Corrales; Ana Belén García Delgado; Aránzazu Díaz Cuenca; Sara Borrego González. COMPOSICIÓN BIOPOLIMÉRICA, PROCEDIMIENTO PARA SU PREPARACIÓN Y USO DE LA MISMA. Application number: ES1641.1483. Priority: Spain. Registration Date: 05-11-2019. Applicants: Fundación Pública Andaluza Progreso y Salud, CSIC.
- Juan Carlos Morales, F. Javier Díaz Corrales, Lourdes Valdés, Ana Belén García Delgado, Adoración Montero. COMPUESTOS ACILADOS PARA EL TRATAMIENTO DE PATOLOGÍAS OCULARES. Application number: ES1641.1252 (WO2018096196A1). Priority: Spain. Registration Date: 23-11-2016. Applicants: Fundación Pública Andaluza Progreso y Salud, CSIC.
- Francisco Díaz Corrales; Berta De La Cerda; Lourdes Valdés; Shomi Bhattacharya; Andrea Pensado; Begoña Siego; Alejandro Sánchez. NANOPARTICULATE SYSTEMS FOR USE IN GENE TRANSFER OR GENE DELIVERY. Application number: EP14382221.1 (WO2015189429). Priority: Spain. Registration Date: 17-12-2015. Applicants: Fundación Pública Andaluza Progreso y Salud, Universidad de Santiago de Compostela.

 Dr. Francisco J. Díaz Corrales
Dr. Francisco J. Díaz Corrales
- Dr. Berta De La Cerda Haynes
- Dr. María del Mar Bonillo Lamolda
- Dr. Estefania Caballano Infantes
- Dr. Álvaro Plaza Reyes
- Dr. Lourdes Valdés Sánchez
- Adoración Montero Sánchez



