
Chromatin integrity and function
Overview
Faithful replication of the complete genome is essential for preventing any loss of genetic information. However, this is not an easy task; in fact, the genetic instability that accompanies tumor progression during early stages is associated with replicative stress. Using the yeast Saccharomyces cerevisiae as living model, and a wide repertoire of techniques in genetic, biochemistry, genomic, and molecular and cellular biology, we are focused on two different aspects associated with replication dynamics that have a direct impact on genome integrity and cell cycle progression: chromatin assembly and tolerance to replicative DNA damage. Our main goal is to get a deeper insight into their mechanistic and regulation, as well as in the consequences for cell fitness of mutations in the genes encoding their components.
Recent Research Highlights
A major source of genetic instability is associated with the encounter of the replication fork with DNA adducts that hinder its advance. In this case, replication fork stability and genome integrity are maintained by a number of error-free and error-prone mechanisms that help the fork to pass through the lesions and to fill in the gaps of single-stranded DNA (ssDNA) generated during the process of fork blockage and lesion bypass. Consequently, this DNA damage tolerance (DDT) response is essential for cell cycle progression, genome integrity, and cancer avoidance. DDT relies on homologous recombination (HR) and translesion synthesis (TLS) mechanisms to fill in the ssDNA gaps generated during passing of the replication fork over DNA lesions in the template. Whereas TLS requires specialized polymerases able to incorporate a dNTP opposite the lesion and is error-prone, HR uses the sister chromatid and is mostly error-free. We have reported that the HR protein Rad52 acts in concert with the TLS machinery to repair MMS and UV light-induced ssDNA gaps through different non-recombinogenic mechanisms. Specifically, Rad52 facilitates the recruitment of the Rad6/Rad18 complex, required for PCNA ubiquitylation and subsequent recruitment of the TLS polymerases. Therefore, Rad52 facilitates the tolerance process not only by HR but also by TLS, providing a novel role for the recombination proteins in maintaining genome integrity (Figure 1) (Cano-Linares et al. 2021)
Our previous work ed that restricting Rad52 expression to G2/M impedes Rad51 from binding to ssDNA lesions and replicative damage repair. Therefore, the recruitment of the recombination proteins to ssDNA lesions during DDT must occur during S phase. This is a substantial mechanistic distinction from double-strand break (DSB)-induced HR, in which Rad51 and Rad52 are recruited to the lesions independently of DNA replication, allowing DSB-induced HR to be completed in cells that only express Rad52 in G2/M. These observations raise important questions about the mechanism that facilitate the recruitment of Rad52 and Rad51 to ssDNA lesions during DDT and what determines that the HR proteins drive the tolerance process to TS or TLS. To understand these questions, we have characterized the regulation and functional interactions between Rad51 and Rad52 with the replicative helicase MCM. We that Rad51 and Rad52 interact physically with the MCM helicases loaded in G1 and spread away of replication origins. These interactions require the kinase activity of Cdc7 and are regulated by the cell cycle stage and DNA damage. Interestingly, these interactions take place at nuclease-insoluble nucleoprotein scaffold characterized by the accumulation of replication and repair factors, including the Cdc7 regulator Dbf4. MCM, Rad51 and Rad52 are released from this scaffold during unperturbed replication; however, in the presence of lesions that hamper the advance of the replication fork they remain bound through mechanisms that require the kinase activity of Cdc7 to assist stressed replication forks by facilitating their advance and the repair of ssDNA lesions through non-HR mechanisms (Figure 2) (Cabello-Lobato 2021). A major future challenge will be to purify and characterize those nuclease-resistant nucleoprotein fraction and determine their relevance in the DNA damage tolerance response.
We have also followed our studies about the roles that chromatin assembly plays in the maintenance of genome integrity. These studies have revealed new functions for the process of histone deposition in genetic instability and transcription. First, we have uncovered an unexpected role for programmed histone depletion in the protection of short telomeres during senescence that is mediated by HR. Interestingly, this mechanism is regulated by the kinase Mec1, the yeast homolog of the tumor suppressor gene ATR, suggesting that this factor prevents genetic instability by controlling histone levels and in turn telomere fusions (Barrientos-Moreno et al. 2018).
The importance of chromatin in transcription have been long established. Remarkably, we have n that cohesins regulate transcription by controlling chromatin structure; specifically, they seem to promote transcription activation by helping the RSC chromatin remodeling complex to generate nucleosome free regions at promoters (Maya et al. Epigenetics & Chromatin 2019). Finally, we have characterized the accumulation of DNA damage in cells lacking the chromatin assembly factor Swr1, and established a connection between actin-related complexes, the nuclear envelope and genome integrity (Morillo-Huesca et al. 2019). These genetic results further support previous observations from our group that support the importance of maintaining strict control over the expression and integrity of chromatin remodeling complexes for genome stability and function.
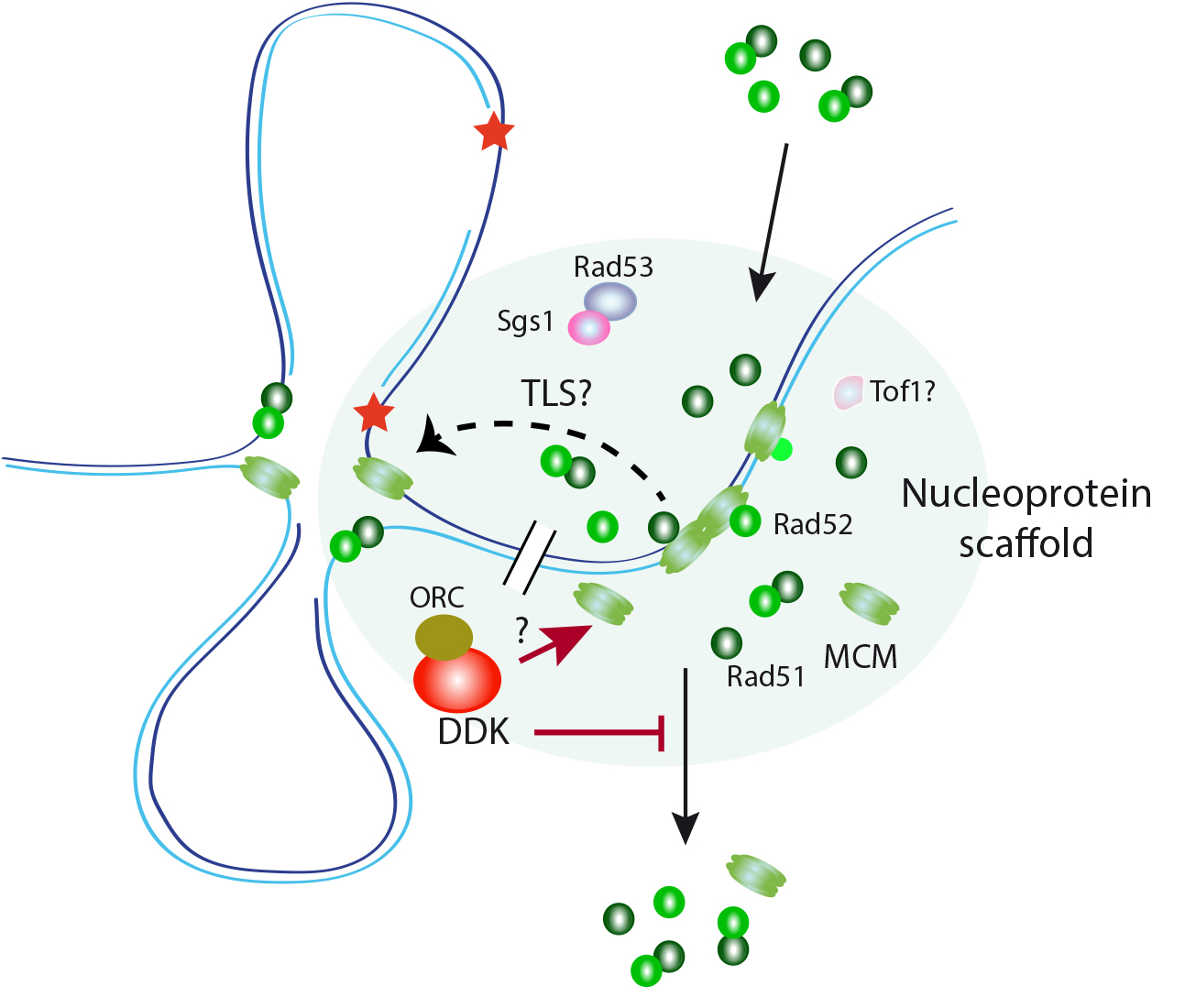
Figure 1. Model for Rad52-mediated tolerance during DDT. The recombination factors Rad52, Rad51 and Rad57 promote efficient Rad6/Rad18 binding to chromatin under unperturbed conditions. In response to blocking lesions that impair replication fork advance the Rad6/Rad18 complex is mobilized to ssDNA lesions where, together with Mms2/Ubc13/Rad5, ubiquitylates PCNA and promotes TS. The ssDNA lesions that escape from this error-free pathway during S phase accumulate in G2/M, where they are filled in by two additional, less accurate mechanisms regulated by Rad52: UbPCNA-independent HR (recombinogenic) and TLS (mutagenic).
Figure 2. A hypothetical role for DDK controlling the integrity of a nucleoprotein scaffold for replication assistance. The MCM complex interacts in yeast with the HR factors Rad51 and Rad52 in a DNA damage and cell cycle-dependent manner. These interactions, which occur at a nuclease-insoluble nucleoprotein scaffold enriched in DNA replication and repair factors, facilitate replication fork progression and ssDNA filling during DDT. DDK maintains these interactions during S phase under conditions of replication stress by preventing the release of these HR factors (and partially of MCM) from the scaffold. DDK might perform this function by controlling either the integrity of the nucleoprotein scaffold by acting upon MCM or the binding of the HR factors to this compartment.
Figure 3. Hypothetical model for gene regulation by cohesins. a Upregulated genes in scc1-73 cells are preferentially in cohesin-enriched regions. Cohesin accumulation might recruit transcription repressors, generate a locally condensed chromatin or hamper the recruitment and/or movement of the transcription machinery. b Downregulated genes in scc1-73 cells are enriched in promoters that have a short or no NFR and a fragile − 1 nucleosome. These promoters require the RSC complex together with additional transcriptional factors to generate a NFR and to activate transcription. Cohesins might collaborate with RSC and other chromatin remodeling factors to remove the nucleosome and/or maintain the NFR.
Selected Publications
- María I. Cano-Linares, Aurora Yáñez-Vilches, Néstor García-Rodríguez, Román González-Prieto, Pedro San-Segundo, Helle D. Ulrich and Félix Prado. Non-recombinogenic role for Rad52, Rad51 and Rad57 in translesion synthesis. EMBO reports (2020) in pressClave: A
- Román Gonzalez-Prieto, María J. Cabello-Lobato and Félix Prado. In vivo binding of recombination proteins to non-DSB DNA lesions and to replication forks. Methods Mol. Biol. (2020) 2153: 447-458 Clave: CL
- Macarena Morillo-Huesca, Marina Murillo-Pineda, Marta Barrientos-Moreno, Elena Gómez-Marín, Marta Clemente-Ruiz and Félix Prado. Actin and nuclear envelope components influence ectopic recombination in the absence of Swr1. Genetics (2019) 213:819-834 Clave: A
- Douglas Maya-Miles, Eloisa Andújar, Monica Pérez-Alegre, Marina Murillo-Pineda, Marta Barrientos-Moreno, María J. Cabello-Lobato, Elena Gómez-Marín, Macarena Morillo-Huesca and Félix Prado. Crosstalk between chromatin structure, cohesion activity and transcription. Epigenetics & Chromatin (2019) 12:47 Clave: A
- Félix Prado. Homologous Recombination: To Fork and Beyond. Genes (2018) 9:603 Clave: R
- Marta Barrientos-Moreno, Marina Murillo Pineda, Ana M. Muñoz-Cabello and Félix Prado. Histone depletion prevents telomere fusions in pre-senescent cells. PLoS Genetics (2018) 14:e1007407 Clave: A
- Félix Prado and Douglas Maya. Regulation of replication fork advance and stability by nucleosome assembly. Genes (2017) 8: 49 Clave: R
- Silvia Jimeno-Gonzalez, Laura Payán, Ana M. Muñoz-Cabello, Macarena Guijo, Gabriel Gutierrez, Félix Prado and José C. Reyes. Chromatin structure regulates RNA polymerase II elongation rate and co-transcriptional pre-mRNA splicing. PNAS (2015) 112:14840-14845 Clave: A
- Marina Murillo-Pineda, María J. Cabello-Lobato, Marta Clemente-Ruíz, Fernando Monje-Casas and Félix Prado. Defective histone supply causes condensin-dependent chromatin alterations, SAC activation and chromosome decatenation impairment. Nucleic Acid Research (2014) 42:12469-12482 Clave: A
- Félix Prado. Genetic instability is prevented by Mrc1-dependent spatio-temporal separation of replicative and repair activities of homologous recombination. BioEssays (2014)36: 451-462 Clave: A
- Román Gonzalez-Prieto, Ana Muñoz-Cabello, María J. Cabello-Lobato and Félix Prado. Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J (2013) 32: 1307-1321 Clave: A
- Marta Clemente-Ruiz, Román Gonzalez-Prieto and Félix Prado. Histone H3K56 acetylation, CAF1 and Rtt106 coordinate nucleosome assembly and stability of advancing replication forks. PloS Genetics (2011) 7: e1002376 Clave: A
- Marta Clemente and Félix Prado. Chromatin assembly controls replication fork stability. EMBO reports (2009) 10: 790-796 Clave: A

 Dr. Felix Prado Velasco
Dr. Felix Prado Velasco
- Dr. Antonia Mª Romero Cuadrado
- Macarena Alcalá Domínguez
- Ana Amiama Roig
- Gabriel Ríos Orelogio



